
PUBLICATION SPOTLIGHT – How a “good” leukemia turns “bad” and how we can turn it “good” again
By Anna S Wilhelmson, Senior Scientist, Porse Group
How would you summarise your study in one sentence?
Through a successful collaboration between the Porse lab here at the Finsen Laboratory and BRIC and the Grebien lab at the University of Veterinary Medicine (Vienna, Austria), we have investigated how mutations in the gene encoding the myeloid transcription factor CEBPA and loss-of-function mutations in the gene encoding the DNA demethylase TET2 cooperate to aggravate acute myeloid leukemia (AML).
Can you describe briefly what you have explored and how?
CEBPA is biallelically mutated (i.e., double mutant; CEBPADM) in 3–15% of AML patients, and N-terminal CEBPA mutations (CEBPANT) promotes the expression of a truncated protein isoform called CEBPA p30. Most patients with CEBPADM AML also feature additional mutations in genes such as GATA2, TET2, WT2, NRAS, FLT3 and CSF3R. For some of these mutations the molecular mechanisms underlying the mutational cooperativity have been identified, however the for the majority this information is lacking, including the loss-of-function mutations in TET2.
In this study we aimed to identify the molecular mechanism underlying cooperation of CEBPADM and TET2MUT. We used transcriptomic and epigenomic analyses of relevant in vitro and in vivo models together with data from AML patients and identified an intricate mechanism where TET2 loss-of-function rebalances GATA2 expression in CEBPADM AML, and thereby drives an aggressive disease.
What would you say is the novelty in this study?
We were able to pinpoint one specific mechanism where loss of TET2 corrects a “weakness” in the CEBPADM AML: CEBPADM AML display increased levels of CEBPA expression compared to that in normal cells. We hypothesized that this would result in increased CEBPA binding to DNA, and since CEBPA can bind TET2, the demethylase would also be recruited to the DNA. We showed that increased CEBPA-TET2 binding at the Gata2 distal hematopoietic enhancer (G2DHE) led to lower DNA methylation at the Gata2 promoter, which resulted in increased expression of Gata2. We further showed that a moderate reduction of Gata2 levels in CebpaDM made the leukemia more aggressive.
Our work therefore shows that when TET2 mutations cooccur with CEBPADM, this suboptimal increase in GATA2 expression is prevented (illustrated in figure 1). Altogether, our work proposes that CEBPA-mutant AMLs acquire additional lesions in genes such as GATA2 and TET2 to reestablish balanced GATA2 levels that permit leukemia development and progression (illustrated in figure 2).
How do the results relate to the scientific field with respect to what is already known?
Mutations in TET2 have been extensively studied, alone and in combination with several other recurrent mutations, and these studies highlights that appropriately regulated DNA methylation is essential for normal hematopoiesis. Despite being highly studied, mechanistic insights of how TET2 mutations cooperates with other types of mutations have been hampered by the fact that malignant cells have been compared to their normal, wild-type counterparts in many of these studies. In the present work, we overcame this limitation by comparing CEBPA-mutant AML in the presence and absence of additional mutations in TET2.
Can you describe the potential of the findings beyond the research field?
We investigated if treatment with the demethylating agent 5-azacytidine would be beneficial in TET2-deficient CEBPA-mutant AML and we were able to show that the proleukemic effect of TET2 mutations can be reversed by 5-azacytidine, including prolonged survival of mice bearing this type of leukemia. Of note, demethylating agents, such as 5-azacytidine, are approved for treatment of AML for patients who are not eligible for hematopoietic stem cell transplantation. This has implications for personalized medicine and suggests that this could be a potential treatment option in CEBPADMTET2MUT patients.
What are the next stages in this research?
Our work shows how a “good “leukemia can turn “bad” and how we can turn it “good” gain, which provide the basis for future research. Even though genetically engineered mouse models to study CEBPA-mutant AML have successfully been used, including in the present study, murine cells are by no means human. Therefore, to better model human AML and yield truly clinical relevance, we are currently studying the combination of CEBPA and TET2 mutations in a human context, utilizing CRISPR technology to genetically engineer primary human hematopoietic stem cells.
Lastly, we hope that our basic research findings may benefit AML patients in a clinical setting, through the use of personalized medicine with better prediction of treatment response based on mutational status or improved targeted therapies.
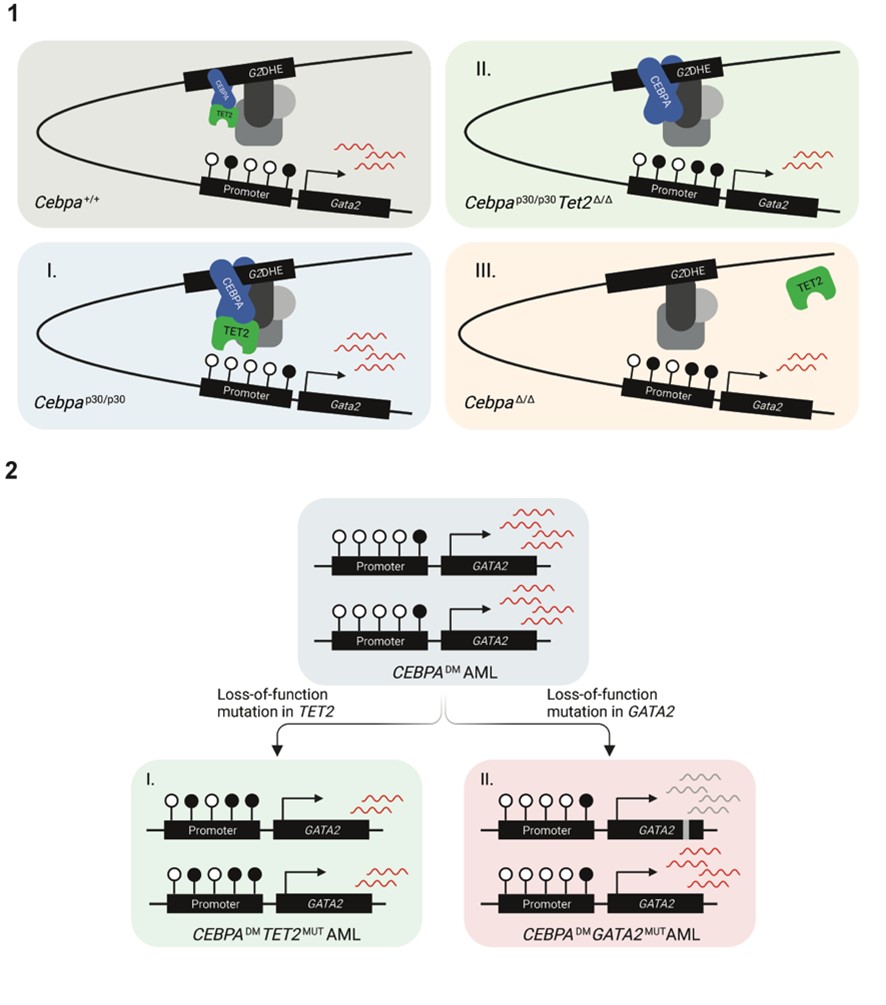
TET2 lesions enhance the aggressiveness of CEBPA-mutant AML by rebalancing GATA2 expression.
1. Model of Gata2 differential expression as a consequence of (I) elevated CEBPA p30 due to the hypermorphic effect of the CEBPANT, (II) TET2 deficiency and, (III) CEBPA deficiency.
2. Schematic illustration of two strategies for CEBPADM AML to rebalance GATA2 levels by (I) loss-of-function mutations in TET2 and (II) loss-of-function mutations in one GATA2 allele.
Read full publication in Nature Communications: TET2 lesions enhance the aggressiveness of CEBPA-mutant acute myeloid leukemia by rebalancing GATA2 expression